Классификация, регрессия и другие алгоритмы Data Mining с использованием R
Использование главных компонент для иерархической кластеризации — один из возможных путей гибридизации алгоритмов, которая все чаще привлекает внимание специалистов. Предварительное сжатие пространства признаков — хороший метод сглаживания случайных флуктуаций в данных, что является предпосылкой построения более стабильных кластерных структур. Это особенно важно для матриц с большим количеством признаков, например, в генной инженерии.
В среде R кластеризация на главные компоненты реализована в пакете FactoMineR и состоит из двух шагов. На первом этапе функцией РСА() выполняется обычный анализ главных компонент и выбирается их число. Далее функция HCPC() использует эти результаты и строит иерархическую кластеризацию. При этом используется метод Уорда, который, как и анализ главных компонент, основан на анализе многомерной дисперсии (inertia).
Опять воспользуемся в качестве примера набором Boston из пакета MASS . Выполним снижение исходной размерности данных, определяемой 14 признаками привлекательности земельных участков Бостона, до 5 главных компонент (рис. 10.13):
library(FactoMineR) library(factoextra) data(Boston, package = "MASS") df.scale scale(Boston) res.pca PCA(df.scale, ncp = 5, graph = TRUE) get_eig(res.pca)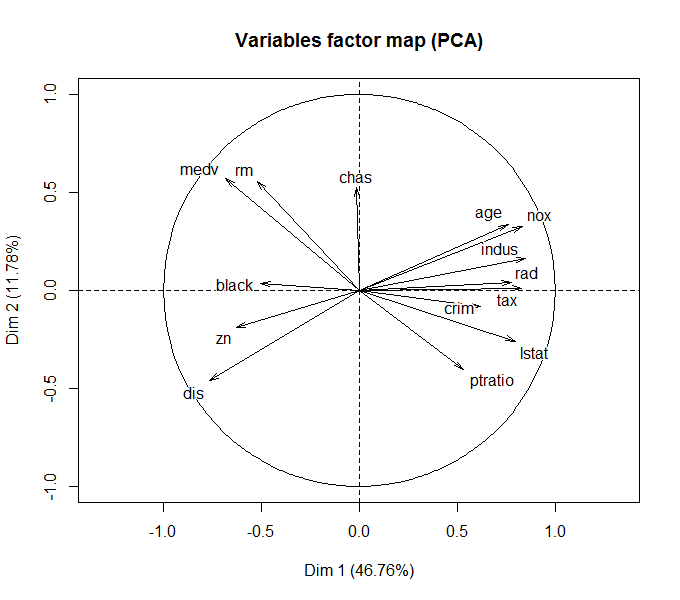
Рисунок 10.13: Разложение исходных признаков по осям двух главных компонент
Напомним, что угол между двумя любыми осями (факторов и/или компонент) на рис. 10.13 соответствует уровню корреляции между ними. Заинтересованный читатель может сравнить этот график с иерархической дендрограммой признаков на рис. 10.12.
Выполним теперь иерархическую кластеризацию на главные компоненты. Построим две диаграммы — обычную дендрограмму, совмещенную с графиком “каменистой осыпи” дисперсии ( choice =»tree» ), и трехмерную дендрограмму, совмещенную с ординационной диаграммой ( choice = «3D.map» ):
res.hcpc HCPC(res.pca, graph = FALSE) plot(res.hcpc, choice = "tree") plot(res.hcpc, choice = "3D.map", ind.names = FALSE)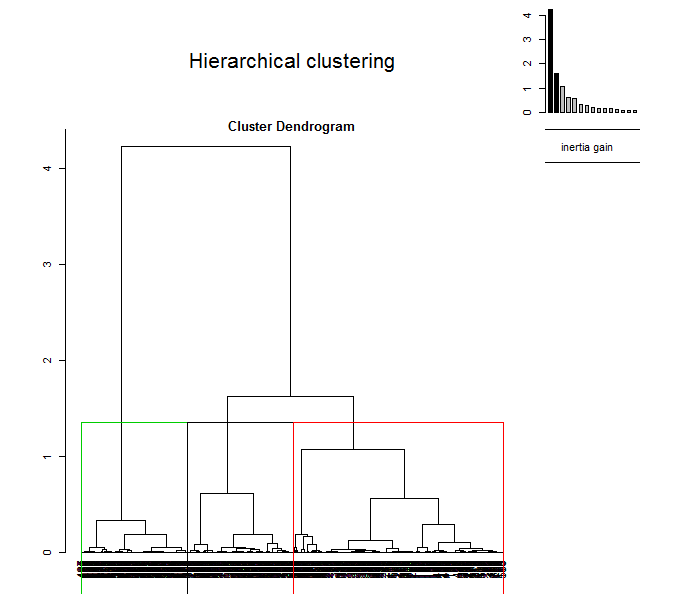
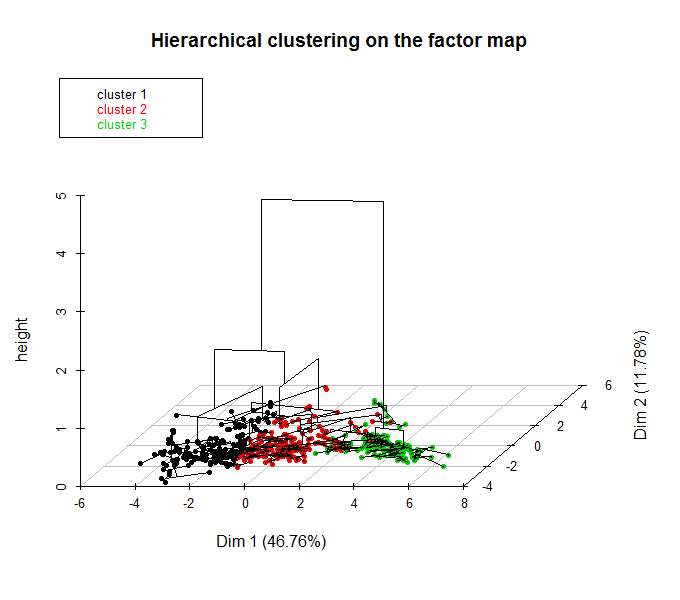
Рисунок 10.14: Дендрограмма с разделением на кластеры (вверху) и гибрид дендрограммы с ординационной диаграммы (внизу)
10.4.2 Метод нечетких k средних (fuzzy analysis clustering)
Традиционные принципы кластерного анализа предполагают, что выделяемые группы представляют собой детерминированные совокупности, т. е. каждый объект может принадлежать только к одному таксону. Ограниченность такого подхода часто приводит к аналитической неопределенности, и поэтому разумной альтернативой понятию абсолютной дискретности является интерпретация компонентов систем как нечетких объектов в составе гибко настраиваемых ординационных структур.
В конце 80-х годов, после того, как основные концепции нечетких множеств (fuzzy sets) были разработаны Лотфи Заде (Zadeh, 1965), началось бурное развитие технических устройств на базе нечетких контроллеров, а нечеткая логика (fuzzy logic) стала неотъемлемой составной частью современных систем искусственного интеллекта.
Множество \(\mathbf\) является нечетким, если существует функция принадлежности (membership function) \(\mu_C(x) = 0\) означает полную несовместимость, т.е. \(x \notin \mathbf\) , а \(\mu_C(x) = 1\) означает полную принадлежность, или \(x \in \mathbf\) . Применительно к кластеризации методом нечетких \(k\) средних функция \(\mu_(\mathbf)\) задает в масштабе от 0 до 1 степень принадлежности каждого объекта \(x_i\) к каждому выделяемому кластеру \(r\) (Bezdek, 1981).
Пусть \(D_ = \sum_j (x_ — \nu_)^2\) — расстояние между каждым \(i\) -м объектом ( \(i = 1, 2, \dots, n\) ), описанным набором признаков \(x_\) , и центрами тяжести \(v_\) каждого из \(k\) кластера ( \(r = 1, 2, \dots, k\) ). Тогда в общем случае кластеризацию объектов \(\mathbf\) можно сформулировать как задачу оптимизации, связанную с нахождением такой матрицы \(\mathbf<\mu>\) , которая минимизировала бы критерий
Экспоненциальный вес ( \(m\) ) в алгоритме нечетких \(k\) средних задает уровень нечеткости получаемых кластеров: чем больше m, тем нечеткое разбиение более “размазано”. “Штатный” диапазон варьирования \(m\) — от 1.2 до 2. Другим важным параметром является количество классов \(k\) , которое принимается из описанных выше соображений.
Выполним построение “нечетких” кластеров для традиционного примера по криминогенной обстановке американских штатов с использованием функции fanny() из пакета cluster :
library(cluster) data("USArrests") set.seed(123) res.fanny fanny(USArrests, k = 4, memb.exp = 1.7, metric = "euclidean", stand = TRUE, maxit = 500) print(head(res.fanny$membership),3)## [,1] [,2] [,3] [,4] ## Alabama 0.639 0.183 0.113 0.0645 ## Alaska 0.337 0.357 0.179 0.1280 ## Arizona 0.213 0.595 0.131 0.0611 ## Arkansas 0.371 0.170 0.264 0.1959 ## California 0.224 0.529 0.160 0.0869 ## Colorado 0.224 0.519 0.174 0.0830res.fanny$coeff## dunn_coeff normalized ## 0.3927126 0.1902834При \(k = 4\) в результате выводится матрица коэффициентов принадлежности, максимальный из которых определяет назначаемый кластер. Так из приведенного фрагмента Алабама и Арканзас включены в кластер 1, а остальные четыре штата — в кластер 2.
Для оценки меры нечеткости полученной классификации используется коэффициент разделения Данна (Dunn):
который принимает минимальное значение при полной нечеткости разбиения, когда расстояния от каждого объекта до центра тяжести любого кластера равновелики: \(\mu = 1/k\) . Напротив, в случае четкой кластеризации ( \(\mu = 1\) или \(\mu = 0\) ) коэффициент Данна \(F_k\) принимает значение 1. Для представленного примера \(F_k = 0.39\) , а его нормированная версия, изменяющаяся от 0 до 1 и характеризующая степень нечеткости \(F_k’ = (kF_k — 1)/(k-1) = 0.19\) .
Построим две диаграммы (рис. 10.15). На первой из них (слева) покажем матрицу \(\sum_^k \mu_^2 / k\) , отсортированную по убыванию (т.е. по степени нечеткости). На второй же (справа) – ординационную диаграмму в пространстве двух главных компонент с результатами кластеризации (аналогична рис. 10.6).
# Визуализация с использованием corrplot library(corrplot) Dunn res.fanny$membership^2 corrplot(Dunn[rev(order(rowSums(Dunn))), ], is.corr = FALSE) # Ординационная диаграмма library(factoextra) fviz_cluster(res.fanny, frame.type = "norm", frame.level = 0.7)
Рисунок 10.15: Матрица степени нечеткости (слева) и ординационная диаграмма (справа), полученные с применением метода нечеткой кластеризации
10.4.3 Статистическая модель кластеризации
Важнейшими свойствами кластеров являются плотность, дисперсия, размер, форма и отделимость (Ким и др., 1989). Характер изменчивости облака экспериментальных точек относительно центроидов искомых кластеров может быть описан статистической моделью со смешанными параметрами (Banfield, Raftery, 1993).
Предположим, что входное множество векторов наблюдений \(x_1, x_2, \dots, x_n\) представляет собой случайные реализации из \(K\) неизвестных распределений \(E_1, E_2, \dots, E_K\) . Допустим, что плотность распределения \(x_i\) , связанная с \(E_k\) , задана функцией \(f_k(x_i, \theta)\) , включающей некоторое неизвестное множество параметров \(\theta\) . Если принять допущение, что классифицируемый объект принадлежит только одному распределению, то можно ввести переменную \(\gamma_I = k\) , если xi принадлежит \(E_k, k = 1, 2, \dots, K\) . Тогда цель оптимизации модели заключается в нахождении таких параметров \(\mathbf\) и \(\mathbf<\gamma>\) , которые максимизируют совокупную вероятность:
Поскольку вероятность \(L(\mathbf)\) основана на смеси \(K\) распределений, то такая модель называется смешанной (mixture model). Если использовать в качестве распределений многомерные нормальные функции, то неизвестные параметры \(\mathbf\) определяются как вектор средних \(\mu_k\) и матрица ковариации \(\Sigma_k\) каждого \(k\) -го распределения.
Если рассматривать изложенный формализм смешанных моделей применительно к кластеризации (model-based clustering), то каждой группе \(k\) соответствует центроид \(\mu_k\) с повышенной плотностью точек в его окрестностях. Геометрические особенности (форма, объем, ориентация) каждого кластера определены матрицей ковариации \(\Sigma_k\) . Главным преимуществом этого подхода, по сравнению с другими методами кластеризации, является возможность автоматической оценки оптимального числа кластеров в процессе подгонки параметров модели.
Выборочные оценки параметров \(\theta\) могут быть получены с использованием алгоритма максимизации математических ожиданий (EM, Expectation-Maximization), который оценивает максимумы вероятности \(L(\mathbf)\) серии моделей: для заданного диапазона значений \(k\) и с различной параметризацией матрицы ковариации. Оптимальная модель обычно отбирается на основе максимума байесовского информационного критерия BIC.
Рассмотрим двумерный набор данных, который содержит время ожидания между извержениями ( waiting ) и продолжительностью извержения ( eruptions ) гейзера “Старый служака” в Йеллоустонском национальном парке. Для иллюстрации характера распределения наблюдаемых данных изобразим диаграмму рассеяния (рис. 10.16):
data(faithful) head(faithful, 3)## eruptions waiting ## 1 3.600 79 ## 2 1.800 54 ## 3 3.333 74library(ggplot2) ggplot(faithful, aes(x = eruptions, y = waiting)) + geom_point() + geom_density2d()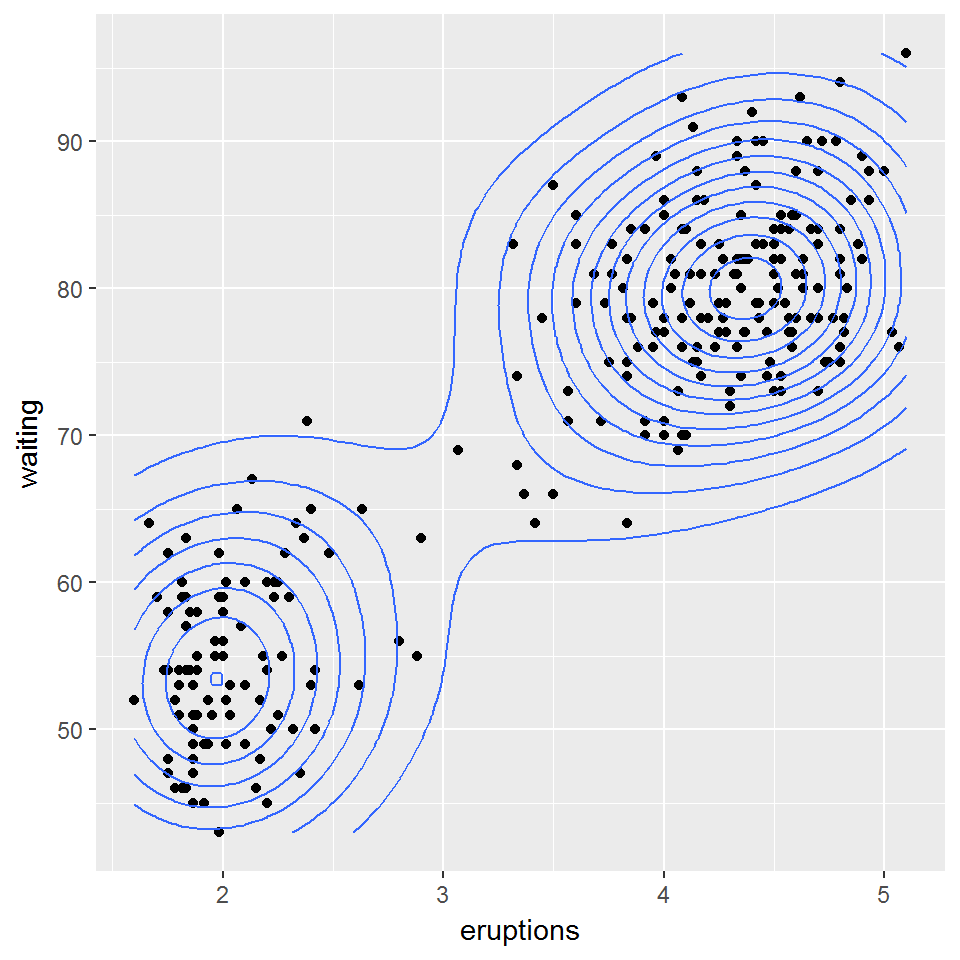
Рисунок 10.16: Диаграмма двумерной плотности распределения данных faithful
Подогнать смешанную модель кластеризации по этим данным можно с использованием функции Mclust() из пакета mclust .
mc mclust::Mclust(faithful) summary(mc)## ---------------------------------------------------- ## Gaussian finite mixture model fitted by EM algorithm ## ---------------------------------------------------- ## ## Mclust EEE (ellipsoidal, equal volume, shape and orientation) model with 3 components: ## ## log.likelihood n df BIC ICL ## -1126.361 272 11 -2314.386 -2360.865 ## ## Clustering table: ## 1 2 3 ## 130 97 45head(mc$z)## [,1] [,2] [,3] ## 1 2.181744e-02 1.130837e-08 9.781825e-01 ## 2 2.475031e-21 1.000000e+00 3.320864e-13 ## 3 2.521625e-03 2.051823e-05 9.974579e-01 ## 4 6.553336e-14 9.999998e-01 1.664978e-07 ## 5 9.838967e-01 7.642900e-20 1.610327e-02 ## 6 2.104355e-07 9.975388e-01 2.461029e-03Мы получили разбиение исходных наблюдений на три кластера с оптимальным BIC = -2314 . Каждому наблюдению назначается кластер с максимальной оцененной вероятностью z . Для визуализации полученных кластеров можно использовать различные варианты функции plot() или fviz_cluster() :
par(mfrow = c(1, 2)) plot(mc, "classification") plot(mc, "uncertainty")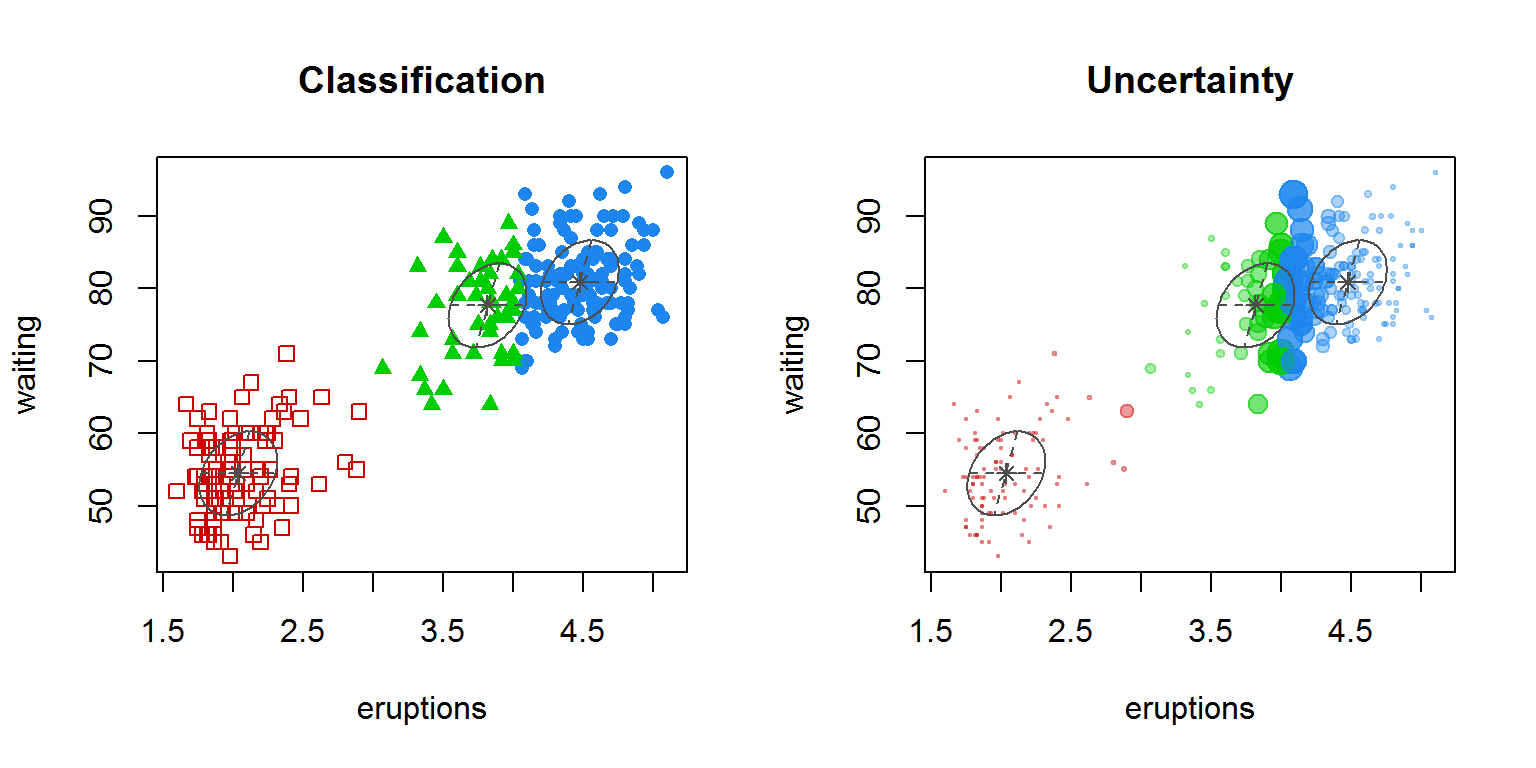
Рисунок 10.17: Распределение наблюдений по кластерам (на графике справа диаметр точек соответствует мере неопределенности — uncertainty )
Как следует из результатов summary() , ковариационные матрицы оптимизированы под вариант структурной организации кластеров EEE (эллипсоидальная с равным объемом, формой и ориентацией). Зависимость критерия BIC от числа кластеров для различных вариантов параметризации можно увидеть на следующем графике:
plot(mc, "BIC")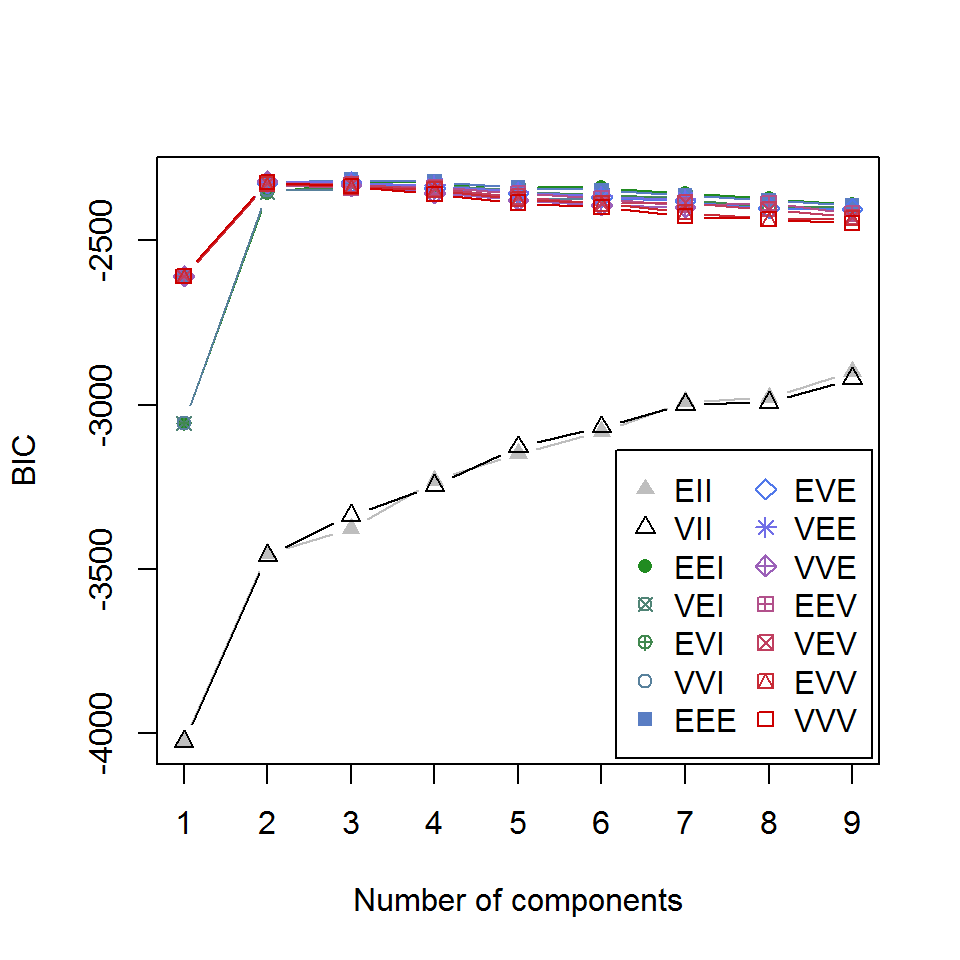
Рисунок 10.18: Зависимость критерия BIC от числа кластеров для различных вариантов параметризации ковариационной матрицы
Каждая опция модели, представленная на рис. 10.18, описана идентификатором, первый символ которого относится к объему, второй — к форме, а третий — к ориентации. Символы могут принимать значения «E» — равный, «V» — переменный и «I» — расположение относительно осей координат. Так, «VEI» означает, что кластеры имеют разный объем, одинаковую форму и одинаково ориентированы по координатным осям. Подробно о составе и смысле опций можно узнать, выполнив команду ?mclustModelNames .
Открытый курс машинного обучения. Тема 7. Обучение без учителя: PCA и кластеризация
Привет всем! Приглашаем изучить седьмую тему нашего открытого курса машинного обучения!

Данное занятие мы посвятим методам обучения без учителя (unsupervised learning), в частности методу главных компонент (PCA — principal component analysis) и кластеризации. Вы узнаете, зачем снижать размерность в данных, как это делать и какие есть способы группирования схожих наблюдений в данных.
UPD 01.2022: С февраля 2022 г. ML-курс ODS на русском возрождается под руководством Петра Ермакова couatl. Для русскоязычной аудитории это предпочтительный вариант (c этими статьями на Хабре – в подкрепление), англоговорящим рекомендуется mlcourse.ai в режиме самостоятельного прохождения.
Видеозапись лекции по мотивам этой статьи в рамках второго запуска открытого курса (сентябрь-ноябрь 2017).
Список статей серии
- Первичный анализ данных с Pandas
- Визуальный анализ данных c Python
- Классификация, деревья решений и метод ближайших соседей
- Линейные модели классификации и регрессии
- Композиции: бэггинг, случайный лес. Кривые валидации и обучения
- Построение и отбор признаков
- Обучение без учителя: PCA, кластеризация
- Обучение на гигабайтах c Vowpal Wabbit
- Анализ временных рядов с помощью Python
- Градиентный бустинг
План этой статьи
- Метод главных компонент (PCA)
- Интуиция, теория и особенности применения
- Примеры использования
- Кластеризация
- K-means
- Affinity Propagation
- Спектральная кластеризация
- Агломеративная кластеризация
- Метрики качества кластеризации
- Домашнее задание
- Полезные источники
0. Введение
Основное отличие методов обучения без учителя от привычных классификаций и регрессий машинного обучения в том, что разметки для данных в этом случае нет. От этого образуются сразу несколько особенностей — во-первых это возможность использования несопоставимо больших объёмов данных, поскольку их не нужно будет размечать руками для обучения, а во-вторых это неясность измерения качества методов, из-за отсутствия таких же прямолинейных и интуитивно понятных метрик, как в задачах обучения с учителем.
Одной из самых очевидных задач, которые возникают в голове в отсутствие явной разметки, является задача снижения размерности данных. С одной стороны её можно рассматривать как помощь в визуализации данных, для этого часто используется метод t-SNE, который мы рассмотрели во второй статье курса. С другой стороны подобное снижение размерности может убрать лишние сильно скоррелированные признаки у наблюдений и подготовить данные для дальнейшей обработки в режиме обучения с учителем, например сделать входные данные более «перевариваемыми» для деревьев решений.
1. Метод главных компонент (PCA)
Интуиция, теория и особенности применения
Метод главных компонент (Principal Component Analysis) — один из самых интуитивно простых и часто используемых методов для снижения размерности данных и проекции их на ортогональное подпространство признаков.

В совсем общем виде это можно представить как предположение о том, что все наши наблюдения скорее всего выглядят как некий эллипсоид в подпространстве нашего исходного пространства и наш новый базис в этом пространстве совпадает с осями этого эллипсоида. Это предположение позволяет нам одновременно избавиться от сильно скоррелированных признаков, так как вектора базиса пространства, на которое мы проецируем, будут ортогональными.
В общем случае размерность этого эллипсоида будет равна размерности исходного пространства, но наше предположение о том, что данные лежат в подпространстве меньшей размерности, позволяет нам отбросить «лишнее» подпространство в новой проекции, а именно то подпространство, вдоль осей которого эллипсоид будет наименее растянут. Мы будем это делать «жадно», выбирая по-очереди в качестве нового элемента базиса нашего нового подпространства последовательно ось эллипсоида из оставшихся, вдоль которой дисперсия будет максимальной.
«To deal with hyper-planes in a 14 dimensional space, visualize a 3D space and say ‘fourteen’ very loudly. Everyone does it.» — Geoffrey Hinton
Рассмотрим как это делается математически:
Чтобы снизить размерность наших данных из в , нам нужно выбрать топ- осей такого эллипсоида, отсортированные по убыванию по дисперсии вдоль осей.
Начнём с того, что посчитаем дисперсии и ковариации исходных признаков. Это делается просто с помощью матрицы ковариации. По определению ковариации, для двух признаков и их ковариация будет
где — матожидание -ого признака.
При этом отметим, что ковариация симметрична и ковариация вектора с самим собой будет равна его дисперсии.
Таким образом матрица ковариации представляет собой симметричную матрицу, где на диагонали лежат дисперсии соответствующих признаков, а вне диагонали — ковариации соответствующих пар признаков. В матричном виде, где это матрица наблюдений, наша матрица ковариации будет выглядеть как
Чтобы освежить память — у матриц как у линейных операторов есть такое интересное свойство как собственные значения и собственные вектора (eigenvalues и eigenvectors). Эти штуки замечательны тем, что когда мы нашей матрицей действуем на соответствующее линейное пространство, собственные вектора остаются на месте и лишь умножаются на соответствующие им собственные значения. То есть определяют подпространство, которое при действии этой матрицей как линейным оператором, остаётся на месте или «переходит в себя». Формально собственный вектор с собственным значением для матрицы определяется просто как .
Матрицу ковариации для нашей выборки можно представить в виде произведения . Из отношения Релея вытекает, что максимальная вариация нашего набора данных будет достигаться вдоль собственного вектора этой матрицы, соответствующего максимальному собственному значению. Таким образом главные компоненты, на которые мы бы хотели спроецировать наши данные, являются просто собственными векторами соответствующих топ- штук собственных значений этой матрицы.
Дальнейшие шаги просты до безобразия — надо просто умножить нашу матрицу данных на эти компоненты и мы получим проекцию наших данных в ортогональном базисе этих компонент. Теперь если мы транспонируем нашу матрицу данных и матрицу векторов главных компонент, мы восстановим исходную выборку в том пространстве, из которого мы делали проекцию на компоненты. Если количество компонент было меньше размерности исходного пространства, мы потеряем часть информации при таком преобразовании.
Примеры использования
Набор данных по цветкам ириса
Начнём с того, что загрузим все необходимые модули и покрутим привычный датасет с ирисами по примеру из документации пакета scikit-learn.
import numpy as np import matplotlib.pyplot as plt import seaborn as sns; sns.set(style='white') %matplotlib inline from sklearn import decomposition from sklearn import datasets from mpl_toolkits.mplot3d import Axes3D # Загрузим наши ириски iris = datasets.load_iris() X = iris.data y = iris.target # Заведём красивую трёхмерную картинку fig = plt.figure(1, figsize=(6, 5)) plt.clf() ax = Axes3D(fig, rect=[0, 0, .95, 1], elev=48, azim=134) plt.cla() for name, label in [('Setosa', 0), ('Versicolour', 1), ('Virginica', 2)]: ax.text3D(X[y == label, 0].mean(), X[y == label, 1].mean() + 1.5, X[y == label, 2].mean(), name, horizontalalignment='center', bbox=dict(alpha=.5, edgecolor='w', facecolor='w')) # Поменяем порядок цветов меток, чтобы они соответствовали правильному y_clr = np.choose(y, [1, 2, 0]).astype(np.float) ax.scatter(X[:, 0], X[:, 1], X[:, 2], c=y_clr, cmap=plt.cm.spectral) ax.w_xaxis.set_ticklabels([]) ax.w_yaxis.set_ticklabels([]) ax.w_zaxis.set_ticklabels([])
Теперь посмотрим, насколько PCA улучшит результаты для модели, которая в данном случае плохо справится с классификацией из-за того, что у неё не хватит сложности для описания данных:
from sklearn.tree import DecisionTreeClassifier from sklearn.model_selection import train_test_split from sklearn.metrics import accuracy_score, roc_auc_score # Выделим из наших данных валидационную выборку X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=.3, stratify=y, random_state=42) # Для примера возьмём неглубокое дерево решений clf = DecisionTreeClassifier(max_depth=2, random_state=42) clf.fit(X_train, y_train) preds = clf.predict_proba(X_test) print('Accuracy: '.format(accuracy_score(y_test, preds.argmax(axis=1))))Out: Accuracy: 0.88889Теперь попробуем сделать то же самое, но с данными, для которых мы снизили размерность до 2D:
# Прогоним встроенный в sklearn PCA pca = decomposition.PCA(n_components=2) X_centered = X - X.mean(axis=0) pca.fit(X_centered) X_pca = pca.transform(X_centered) # И нарисуем получившиеся точки в нашем новом пространстве plt.plot(X_pca[y == 0, 0], X_pca[y == 0, 1], 'bo', label='Setosa') plt.plot(X_pca[y == 1, 0], X_pca[y == 1, 1], 'go', label='Versicolour') plt.plot(X_pca[y == 2, 0], X_pca[y == 2, 1], 'ro', label='Virginica') plt.legend(loc=0);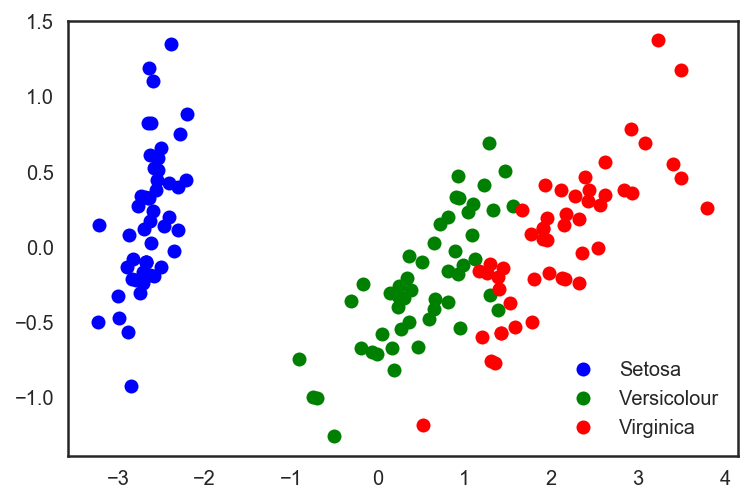
# Повторим то же самое разбиение на валидацию и тренировочную выборку. X_train, X_test, y_train, y_test = train_test_split(X_pca, y, test_size=.3, stratify=y, random_state=42) clf = DecisionTreeClassifier(max_depth=2, random_state=42) clf.fit(X_train, y_train) preds = clf.predict_proba(X_test) print('Accuracy: '.format(accuracy_score(y_test, preds.argmax(axis=1))))Смотрим на возросшую точность классификации:
Out: Accuracy: 0.91111Видно, что качество возросло незначительно, но для более сложных данных более высокой размерности, где данные не разбиваются тривиально вдоль одного признака, применение PCA может достаточно сильно улучшить качество работы деревьев решений и ансамблей на их основе.
Посмотрим на 2 главные компоненты в последнем PCA-представлении данных и на тот процент исходной дисперсии в даных, который они «объясняют».
for i, component in enumerate(pca.components_): print("<> component: <>% of initial variance".format(i + 1, round(100 * pca.explained_variance_ratio_[i], 2))) print(" + ".join("%.3f x %s" % (value, name) for value, name in zip(component, iris.feature_names)))1 component: 92.46% of initial variance 0.362 x sepal length (cm) + -0.082 x sepal width (cm) + 0.857 x petal length (cm) + 0.359 x petal width (cm) 2 component: 5.3% of initial variance 0.657 x sepal length (cm) + 0.730 x sepal width (cm) + -0.176 x petal length (cm) + -0.075 x petal width (cm)Набор данных по рукописным цифрам
Теперь возьмем набор данных по рукописным цифрам. Мы с ним уже работали в 3 статье про деревья решений и метод ближайших соседей.
digits = datasets.load_digits() X = digits.data y = digits.targetВспомним, как выглядят эти цифры – посмотрим на первые десять. Картинки здесь представляются матрицей 8 x 8 (интенсивности белого цвета для каждого пикселя). Далее эта матрица «разворачивается» в вектор длины 64, получается признаковое описание объекта.
# f, axes = plt.subplots(5, 2, sharey=True, figsize=(16,6)) plt.figure(figsize=(16, 6)) for i in range(10): plt.subplot(2, 5, i + 1) plt.imshow(X[i,:].reshape([8,8]));
Получается, размерность признакового пространства здесь – 64. Но давайте снизим размерность всего до 2 и увидим, что даже на глаз рукописные цифры неплохо разделяются на кластеры.
pca = decomposition.PCA(n_components=2) X_reduced = pca.fit_transform(X) print('Projecting %d-dimensional data to 2D' % X.shape[1]) plt.figure(figsize=(12,10)) plt.scatter(X_reduced[:, 0], X_reduced[:, 1], c=y, edgecolor='none', alpha=0.7, s=40, cmap=plt.cm.get_cmap('nipy_spectral', 10)) plt.colorbar() plt.title('MNIST. PCA projection')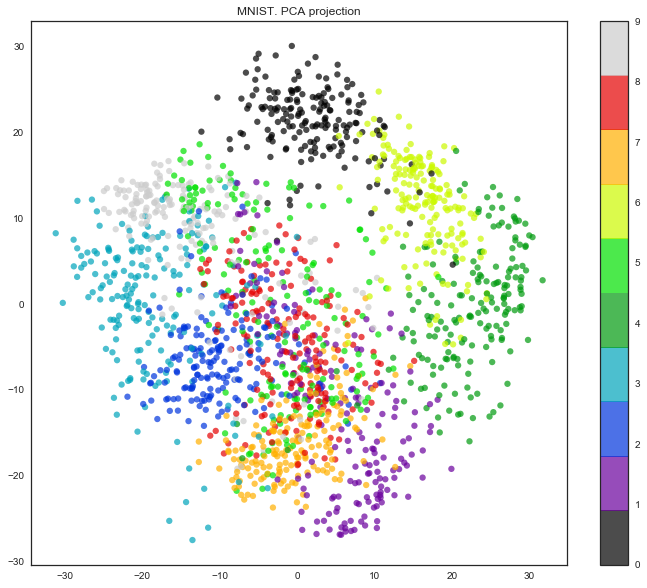
Ну, правда, с t-SNE картинка получается еще лучше, поскольку у PCA ограничение – он находит только линейные комбинации исходных признаков. Зато даже на этом относительно небольшом наборе данных можно заметить, насколько t-SNE дольше работает.
%%time from sklearn.manifold import TSNE tsne = TSNE(random_state=17) X_tsne = tsne.fit_transform(X) plt.figure(figsize=(12,10)) plt.scatter(X_tsne[:, 0], X_tsne[:, 1], c=y, edgecolor='none', alpha=0.7, s=40, cmap=plt.cm.get_cmap('nipy_spectral', 10)) plt.colorbar() plt.title('MNIST. t-SNE projection')
На практике, как правило, выбирают столько главных компонент, чтобы оставить 90% дисперсии исходных данных. В данном случае для этого достаточно выделить 21 главную компоненту, то есть снизить размерность с 64 признаков до 21.
pca = decomposition.PCA().fit(X) plt.figure(figsize=(10,7)) plt.plot(np.cumsum(pca.explained_variance_ratio_), color='k', lw=2) plt.xlabel('Number of components') plt.ylabel('Total explained variance') plt.xlim(0, 63) plt.yticks(np.arange(0, 1.1, 0.1)) plt.axvline(21, c='b') plt.axhline(0.9, c='r') plt.show();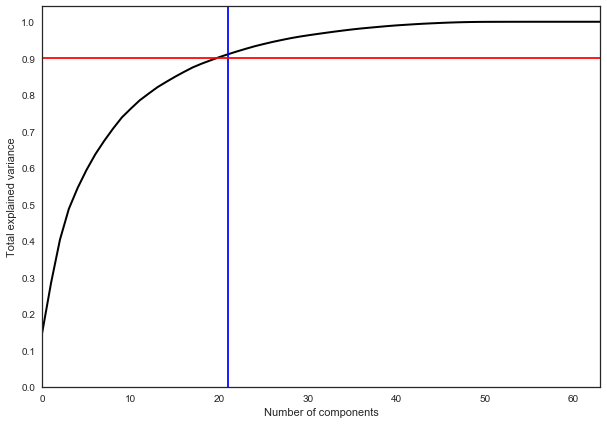
2. Кластеризация
Интуитивная постановка задачи кластеризации довольно проста и представляет из себя наше желание сказать: «Вот тут у меня насыпаны точки. Я вижу, что они сваливаются в какие-то кучки вместе. Было бы круто иметь возможность эти точки относить к кучкам и в случае появления новой точки на плоскости говорить, в какую кучку она падает.» Из такой постановки видно, что пространства для фантазии получается много, и от этого возникает соответствующее множество алгоритмов решения этой задачи. Перечисленные алгоритмы ни в коем случае не описывают данное множество полностью, но являются примерами самых популярных методов решения задачи кластеризации.
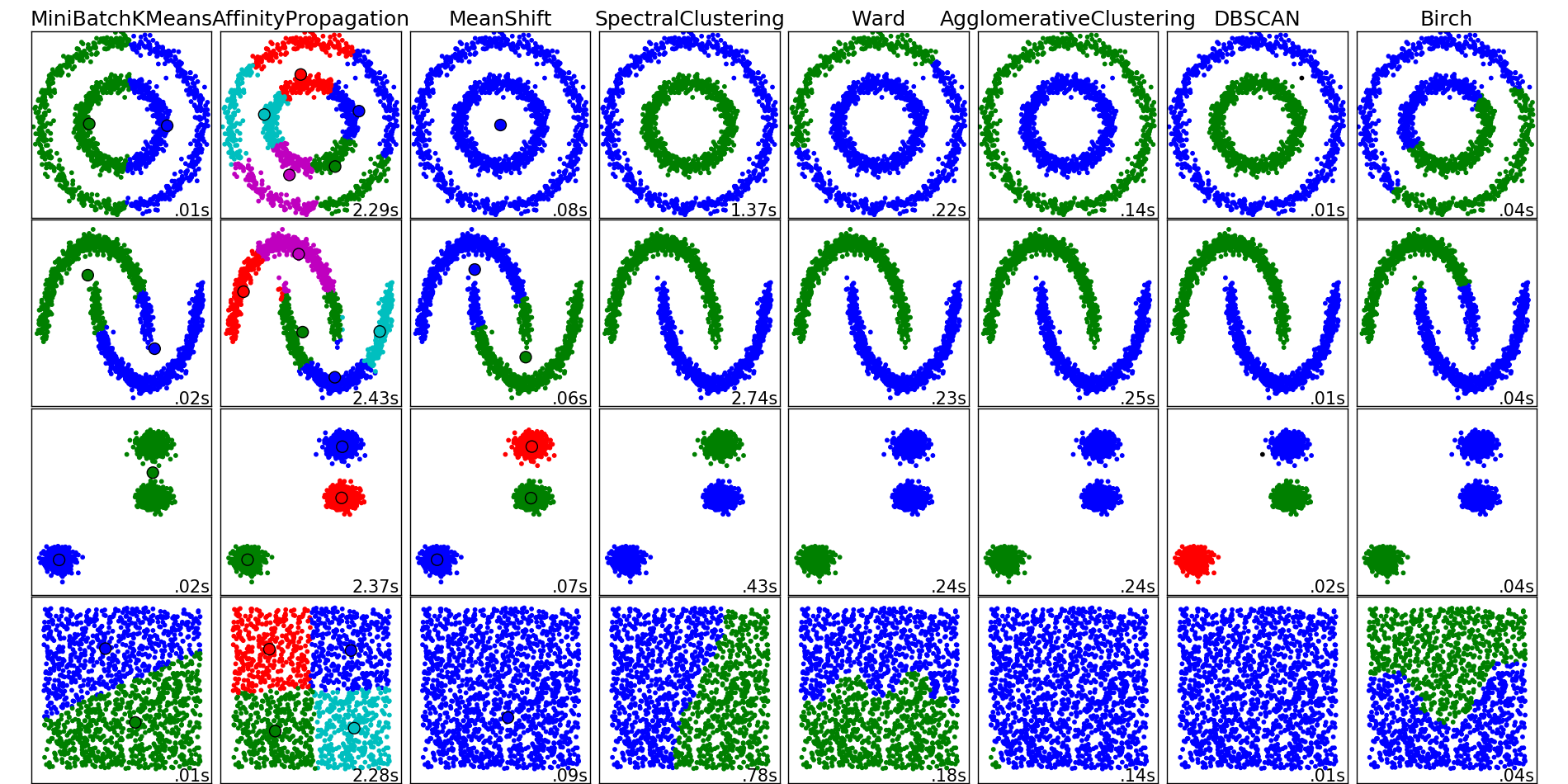
Примеры работы алгоритмов кластеризации из документации пакета scikit-learn
K-means
Алгоритм К-средних, наверное, самый популярный и простой алгоритм кластеризации и очень легко представляется в виде простого псевдокода:
- Выбрать количество кластеров , которое нам кажется оптимальным для наших данных.
- Высыпать случайным образом в пространство наших данных точек (центроидов).
- Для каждой точки нашего набора данных посчитать, к какому центроиду она ближе.
- Переместить каждый центроид в центр выборки, которую мы отнесли к этому центроиду.
- Повторять последние два шага фиксированное число раз, либо до тех пор пока центроиды не «сойдутся» (обычно это значит, что их смещение относительно предыдущего положения не превышает какого-то заранее заданного небольшого значения).
В случае обычной евклидовой метрики для точек лежащих на плоскости, этот алгоритм очень просто расписывается аналитически и рисуется. Давайте посмотрим соответствующий пример:
# Начнём с того, что насыпем на плоскость три кластера точек X = np.zeros((150, 2)) np.random.seed(seed=42) X[:50, 0] = np.random.normal(loc=0.0, scale=.3, size=50) X[:50, 1] = np.random.normal(loc=0.0, scale=.3, size=50) X[50:100, 0] = np.random.normal(loc=2.0, scale=.5, size=50) X[50:100, 1] = np.random.normal(loc=-1.0, scale=.2, size=50) X[100:150, 0] = np.random.normal(loc=-1.0, scale=.2, size=50) X[100:150, 1] = np.random.normal(loc=2.0, scale=.5, size=50) plt.figure(figsize=(5, 5)) plt.plot(X[:, 0], X[:, 1], 'bo');
# В scipy есть замечательная функция, которая считает расстояния # между парами точек из двух массивов, подающихся ей на вход from scipy.spatial.distance import cdist # Прибьём рандомность и насыпем три случайные центроиды для начала np.random.seed(seed=42) centroids = np.random.normal(loc=0.0, scale=1., size=6) centroids = centroids.reshape((3, 2)) cent_history = [] cent_history.append(centroids) for i in range(3): # Считаем расстояния от наблюдений до центроид distances = cdist(X, centroids) # Смотрим, до какой центроиде каждой точке ближе всего labels = distances.argmin(axis=1) # Положим в каждую новую центроиду геометрический центр её точек centroids = centroids.copy() centroids[0, :] = np.mean(X[labels == 0, :], axis=0) centroids[1, :] = np.mean(X[labels == 1, :], axis=0) centroids[2, :] = np.mean(X[labels == 2, :], axis=0) cent_history.append(centroids)# А теперь нарисуем всю эту красоту plt.figure(figsize=(8, 8)) for i in range(4): distances = cdist(X, cent_history[i]) labels = distances.argmin(axis=1) plt.subplot(2, 2, i + 1) plt.plot(X[labels == 0, 0], X[labels == 0, 1], 'bo', label='cluster #1') plt.plot(X[labels == 1, 0], X[labels == 1, 1], 'co', label='cluster #2') plt.plot(X[labels == 2, 0], X[labels == 2, 1], 'mo', label='cluster #3') plt.plot(cent_history[i][:, 0], cent_history[i][:, 1], 'rX') plt.legend(loc=0) plt.title('Step '.format(i + 1));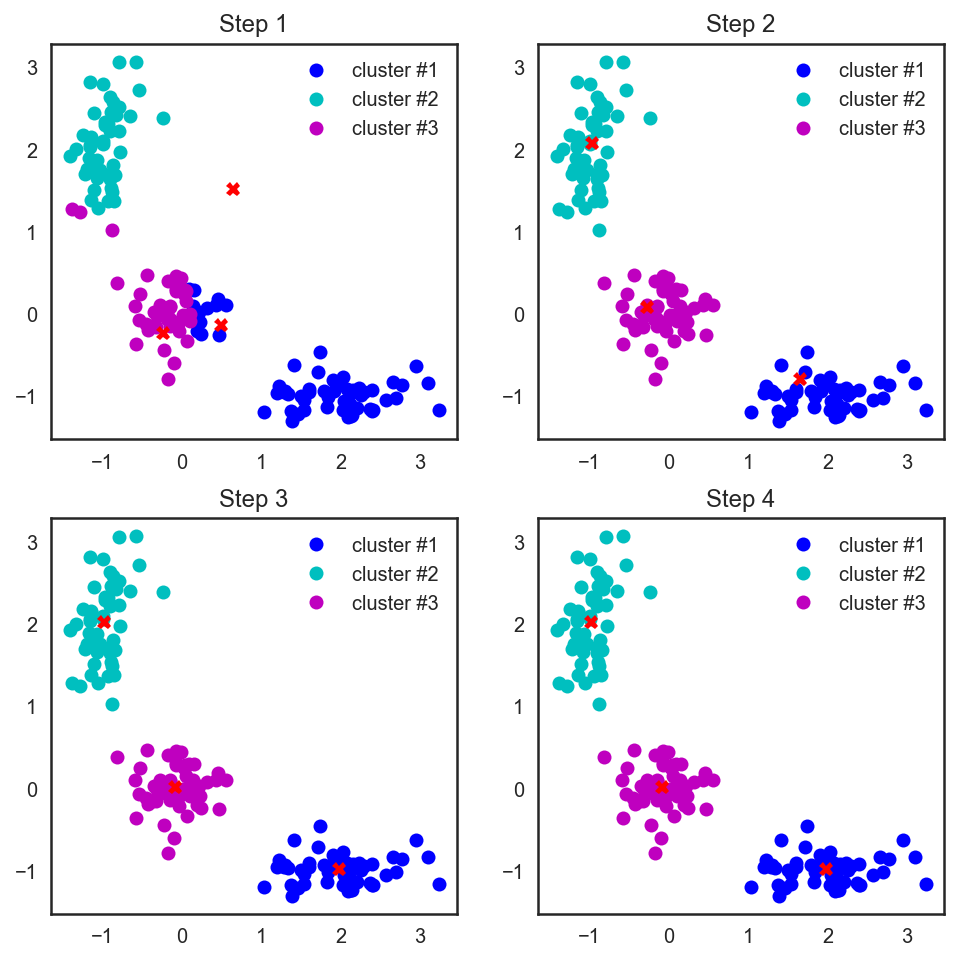
Также стоит заметить, что хоть мы и рассматривали евклидово расстояние, алгоритм будет сходиться и в случае любой другой метрики, поэтому для различных задач кластеризации в зависимости от данных можно экспериментировать не только с количеством шагов или критерием сходимости, но и с метрикой, по которой мы считаем расстояния между точками и центроидами кластеров.
Другой особенностью этого алгоритма является то, что он чувствителен к исходному положению центроид кластеров в пространстве. В такой ситуации спасает несколько последовательных запусков алгоритма с последующим усреднением полученных кластеров.
Выбор числа кластеров для kMeans
В отличие от задачи классификации или регресии, в случае кластеризации сложнее выбрать критерий, с помощью которого было бы просто представить задачу кластеризации как задачу оптимизации.
В случае kMeans распространен вот такой критерий – сумма квадратов расстояний от точек до центроидов кластеров, к которым они относятся.
здесь – множество кластеров мощности , – центроид кластера .
Понятно, что здравый смысл в этом есть: мы хотим, чтобы точки располагались кучно возле центров своих кластеров. Но вот незадача: минимум такого функционала будет достигаться тогда, когда кластеров столько же, сколько и точек (то есть каждая точка – это кластер из одного элемента).
Для решения этого вопроса (выбора числа кластеров) часто пользуются такой эвристикой: выбирают то число кластеров, начиная с которого описанный функционал падает «уже не так быстро». Или более формально:
from sklearn.cluster import KMeans inertia = [] for k in range(1, 8): kmeans = KMeans(n_clusters=k, random_state=1).fit(X) inertia.append(np.sqrt(kmeans.inertia_)) plt.plot(range(1, 8), inertia, marker='s'); plt.xlabel('$k$') plt.ylabel('$J(C_k)$');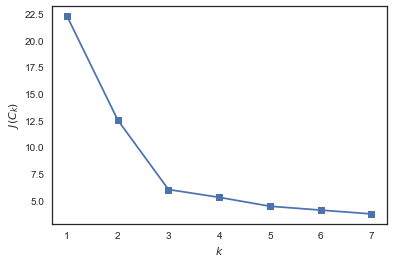
Видим, что падает сильно при увеличении числа кластеров с 1 до 2 и с 2 до 3 и уже не так сильно – при изменении с 3 до 4. Значит, в данной задаче оптимально задать 3 кластера.
Сложности
Само по себе решение задачи K-means NP-трудное (NP-hard), и для размерности , числа кластеров и числа точек решается за . Для решения такой боли часто используются эвристики, например MiniBatch K-means, который для обучения использует не весь датасет целиком, а лишь маленькие его порции (batch) и обновляет центроиды используя среднее за всю историю обновлений центроида от всех относящихся к нему точек. Сравнение обычного K-means и его MiniBatch имплементации можно посмотреть в документации scikit-learn.
Реализация алгоритма в scikit-learn обладает массой удобных плюшек, таких как возможность задать количество запусков через параметр n_init , что даст более устойчивые центроиды для кластеров в случае скошенных данных. К тому же эти запуски можно делать параллельно, не жертвуя временем вычисления.
Affinity Propagation
Ещё один пример алгоритма кластеризации. В отличие от алгоритма К-средних, данный подход не требует заранее определять число кластеров, на которое мы хотим разбить наши данные. Основная идея алгоритма заключается в том, что нам хотелось бы, чтобы наши наблюдения кластеризовались в группы на основе того, как они «общаются», или насколько они похожи друг на друга.
Заведём для этого какую-нибудь метрику «похожести», определяющуюся тем, что если наблюдение больше похоже на наблюдение , чем на . Простым примером такой похожести будет отрицательный квадрат расстояния .
Теперь опишем сам процесс «общения». Для этого заведём две матрицы, инициализируемые нулями, одна из которых будет описывать, насколько хорошо -тое наблюдение подходит для того, чтобы быть «примером для подражания» для -того наблюдения относительно всех остальных потенциальных «примеров», а вторая — будет описывать, насколько правильным было бы для -того наблюдения выбрать -тое в качестве такого «примера». Звучит немного запутанно, но чуть дальше увидим пример «на пальцах».
После этого данные матрицы обновляются по очереди по правилам:
Спектральная кластеризация
Спектральная кластеризация объединяет несколько описанных выше подходов, чтобы получить максимальное количество профита от сложных многообразий размерности меньшей исходного пространства.
Для работы этого алгоритма нам потребуется определить матрицу похожести наблюдений (adjacency matrix). Можно это сделать таким же образом, как и для Affinity Propagation выше: . Эта матрица также описывает полный граф с вершинами в наших наблюдениях и рёбрами между каждой парой наблюдений с весом, соответствующим степени похожести этих вершин. Для нашей выше выбранной метрики и точек, лежащих на плоскости, эта штука будет интуитивной и простой — две точки более похожи, если ребро между ними короче. Теперь нам бы хотелось разделить наш получившийся граф на две части так, чтобы получившиеся точки в двух графах были в общем больше похожи на другие точки внутри получившейся «своей» половины графа, чем на точки в «другой» половине. Формальное название такой задачи называется Normalized cuts problem и подробнее про это можно почитать тут.
Агломеративная кластеризация
Наверное самый простой и понятный алгоритм кластеризации без фиксированного числа кластеров — агломеративная кластеризация. Интуиция у алгоритма очень простая:
- Начинаем с того, что высыпаем на каждую точку свой кластер
- Сортируем попарные расстояния между центрами кластеров по возрастанию
- Берём пару ближайших кластеров, склеиваем их в один и пересчитываем центр кластера
- Повторяем п. 2 и 3 до тех пор, пока все данные не склеятся в один кластер
Сам процесс поиска ближайших кластеров может происходить с использованием разных методов объединения точек:
- Single linkage — минимум попарных расстояний между точками из двух кластеров
- Complete linkage — максимум попарных расстояний между точками из двух кластеров
- Average linkage — среднее попарных расстояний между точками из двух кластеров
- Centroid linkage — расстояние между центроидами двух кластеров
Профит первых трёх подходов по сравнению с четвёртым в том, что для них не нужно будет пересчитывать расстояния каждый раз после склеивания, что сильно снижает вычислительную сложность алгоритма.
По итогам выполнения такого алгоритма можно также построить замечательное дерево склеивания кластеров и глядя на него определить, на каком этапе нам было бы оптимальнее всего остановить алгоритм. Либо воспользоваться тем же правилом локтя, что и в k-means.
К счастью для нас в питоне уже есть замечательные инструменты для построения таких дендрограмм для агломеративной кластеризации. Рассмотрим на примере наших кластеров из K-means:
from scipy.cluster import hierarchy from scipy.spatial.distance import pdist X = np.zeros((150, 2)) np.random.seed(seed=42) X[:50, 0] = np.random.normal(loc=0.0, scale=.3, size=50) X[:50, 1] = np.random.normal(loc=0.0, scale=.3, size=50) X[50:100, 0] = np.random.normal(loc=2.0, scale=.5, size=50) X[50:100, 1] = np.random.normal(loc=-1.0, scale=.2, size=50) X[100:150, 0] = np.random.normal(loc=-1.0, scale=.2, size=50) X[100:150, 1] = np.random.normal(loc=2.0, scale=.5, size=50) distance_mat = pdist(X) # pdist посчитает нам верхний треугольник матрицы попарных расстояний Z = hierarchy.linkage(distance_mat, 'single') # linkage — реализация агломеративного алгоритма plt.figure(figsize=(10, 5)) dn = hierarchy.dendrogram(Z, color_threshold=0.5)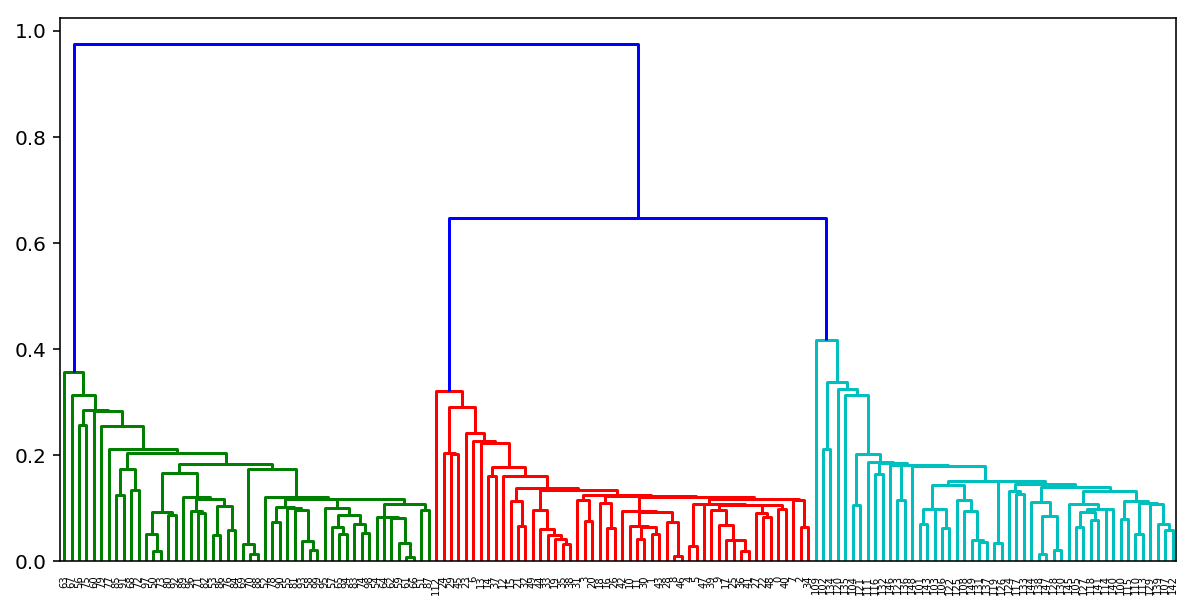
Метрики качества кластеризации
Задача оценки качества кластеризации является более сложной по сравнению с оценкой качества классификации. Во-первых, такие оценки не должны зависеть от самих значений меток, а только от самого разбиения выборки. Во-вторых, не всегда известны истинные метки объектов, поэтому также нужны оценки, позволяющие оценить качество кластеризации, используя только неразмеченную выборку.
Выделяют внешние и внутренние метрики качества. Внешние используют информацию об истинном разбиении на кластеры, в то время как внутренние метрики не используют никакой внешней информации и оценивают качество кластеризации, основываясь только на наборе данных. Оптимальное число кластеров обычно определяют с использованием внутренних метрик.
Все указанные ниже метрики реализованы в sklearn.metrics .
Adjusted Rand Index (ARI)
Предполагается, что известны истинные метки объектов. Данная мера не зависит от самих значений меток, а только от разбиения выборки на кластеры. Пусть — число объектов в выборке. Обозначим через — число пар объектов, имеющих одинаковые метки и находящихся в одном кластере, через — число пар объектов, имеющих различные метки и находящихся в разных кластерах. Тогда Rand Index это
То есть это доля объектов, для которых эти разбиения (исходное и полученное в результате кластеризации) «согласованы». Rand Index (RI) выражает схожесть двух разных кластеризаций одной и той же выборки. Чтобы этот индекс давал значения близкие к нулю для случайных кластеризаций при любом и числе кластеров, необходимо нормировать его. Так определяется Adjusted Rand Index:
Эта мера симметрична, не зависит от значений и перестановок меток. Таким образом, данный индекс является мерой расстояния между различными разбиениями выборки. принимает значения в диапазоне . Отрицательные значения соответствуют «независимым» разбиениям на кластеры, значения, близкие к нулю, — случайным разбиениям, и положительные значения говорят о том, что два разбиения схожи (совпадают при ).
Adjusted Mutual Information (AMI)
Данная мера очень похожа на . Она также симметрична, не зависит от значений и перестановок меток. Определяется с использованием функции энтропии, интерпретируя разбиения выборки, как дискретные распределения (вероятность отнесения к кластеру равна доле объектов в нём). Индекс определяется как взаимная информация для двух распределений, соответствующих разбиениям выборки на кластеры. Интуитивно, взаимная информация измеряет долю информации, общей для обоих разбиений: насколько информация об одном из них уменьшает неопределенность относительно другого.
Аналогично определяется индекс , позволяющий избавиться от роста индекса с увеличением числа классов. Он принимает значения в диапазоне . Значения, близкие к нулю, говорят о независимости разбиений, а близкие к единице – об их схожести (совпадении при ).
Гомогенность, полнота, V-мера
Формально данные меры также определяются с использованием функций энтропии и условной энтропии, рассматривая разбиения выборки как дискретные распределения:
здесь — результат кластеризации, — истинное разбиение выборки на классы. Таким образом, измеряет, насколько каждый кластер состоит из объектов одного класса, а — насколько объекты одного класса относятся к одному кластеру. Эти меры не являются симметричными. Обе величины принимают значения в диапазоне , и большие значения соответствуют более точной кластеризации. Эти меры не являются нормализованными, как или , и поэтому зависят от числа кластеров. Случайная кластеризация не будет давать нулевые показатели при большом числе классов и малом числе объектов. В этих случаях предпочтительнее использовать . Однако при числе объектов более 1000 и числе кластеров менее 10 данная проблема не так явно выражена и может быть проигнорирована.
Для учёта обеих величин и одновременно вводится -мера, как их среднее гармоническое:
Она является симметричной и показывает, насколько две кластеризации схожи между собой.
Силуэт
В отличие от описанных выше метрик, данный коэффициент не предполагает знания истинных меток объектов, и позволяет оценить качество кластеризации, используя только саму (неразмеченную) выборку и результат кластеризации. Сначала силуэт определяется отдельно для каждого объекта. Обозначим через — среднее расстояние от данного объекта до объектов из того же кластера, через — среднее расстояние от данного объекта до объектов из ближайшего кластера (отличного от того, в котором лежит сам объект). Тогда силуэтом данного объекта называется величина:
Силуэтом выборки называется средняя величина силуэта объектов данной выборки. Таким образом, силуэт показывает, насколько среднее расстояние до объектов своего кластера отличается от среднего расстояния до объектов других кластеров. Данная величина лежит в диапазоне . Значения, близкие к -1, соответствуют плохим (разрозненным) кластеризациям, значения, близкие к нулю, говорят о том, что кластеры пересекаются и накладываются друг на друга, значения, близкие к 1, соответствуют «плотным» четко выделенным кластерам. Таким образом, чем больше силуэт, тем более четко выделены кластеры, и они представляют собой компактные, плотно сгруппированные облака точек.
С помощью силуэта можно выбирать оптимальное число кластеров (если оно заранее неизвестно) — выбирается число кластеров, максимизирующее значение силуэта. В отличие от предыдущих метрик, силуэт зависит от формы кластеров, и достигает больших значений на более выпуклых кластерах, получаемых с помощью алгоритмов, основанных на восстановлении плотности распределения.
И напоследок давайте посмотрим на эти метрики для наших алгоритмов, запущенных на данных рукописных цифр MNIST:
from sklearn import metrics from sklearn import datasets import pandas as pd from sklearn.cluster import KMeans, AgglomerativeClustering, AffinityPropagation, SpectralClustering data = datasets.load_digits() X, y = data.data, data.target algorithms = [] algorithms.append(KMeans(n_clusters=10, random_state=1)) algorithms.append(AffinityPropagation()) algorithms.append(SpectralClustering(n_clusters=10, random_state=1, affinity='nearest_neighbors')) algorithms.append(AgglomerativeClustering(n_clusters=10)) data = [] for algo in algorithms: algo.fit(X) data.append((< 'ARI': metrics.adjusted_rand_score(y, algo.labels_), 'AMI': metrics.adjusted_mutual_info_score(y, algo.labels_), 'Homogenity': metrics.homogeneity_score(y, algo.labels_), 'Completeness': metrics.completeness_score(y, algo.labels_), 'V-measure': metrics.v_measure_score(y, algo.labels_), 'Silhouette': metrics.silhouette_score(X, algo.labels_)>)) results = pd.DataFrame(data=data, columns=['ARI', 'AMI', 'Homogenity', 'Completeness', 'V-measure', 'Silhouette'], index=['K-means', 'Affinity', 'Spectral', 'Agglomerative']) results| ARI | AMI | Homogenity | Completeness | V-measure | Silhouette | |
|---|---|---|---|---|---|---|
| K-means | 0.662295 | 0.732799 | 0.735448 | 0.742972 | 0.739191 | 0.182097 |
| Affinity | 0.175174 | 0.451249 | 0.958907 | 0.486901 | 0.645857 | 0.115197 |
| Spectral | 0.752639 | 0.827818 | 0.829544 | 0.876367 | 0.852313 | 0.182195 |
| Agglomerative | 0.794003 | 0.856085 | 0.857513 | 0.879096 | 0.868170 | 0.178497 |
3. Домашнее задание
В демо-версии домашнего задания предлагается поработать с данными Samsung по распознаванию видов активностей людей. Задача интересная, мы на нее посмотрим и как на задачу кластеризации (забыв, что выборка размечена) и как на задачу классификации. Jupyter-заготовка, веб-форма для ответов, там же найдете и решение.
Актуальные и обновляемые версии демо-заданий – на английском на сайте курса, вот первое задание. Также по подписке на Patreon («Bonus Assignments» tier) доступны расширенные домашние задания по каждой теме (только на англ.).
4. Полезные источники
- Open Machine Learning Course. Topic 7. Unsupervised Learning: PCA and Clustering
- Видеозапись лекции по мотивам этой статьи
- Перевод этой статьи на английский – Medium story
- статья «Как подобрать платье с помощью метода главных компонент»
- статья «Как работает метод главных компонент (PCA) на простом примере»
- статья «Интересные алгоритмы кластеризации, часть первая: Affinity propagation»
- статья «Интересные алгоритмы кластеризации, часть вторая: DBSCAN»
- обзор методов кластеризации в пакете scikit-learn (англ.)
- Q&A Разбор PCA с интуицией и примерами (англ.)
- тетрадка про k-Means и EM-алгоритм в курсе Дмитрия Ефимова (англ.)
- конспект «Обучение без учителя» в курсе Евгения Соколова
Статья написана в соавторстве с yorko (Юрием Кашницким). Материал статьи доступен в виде тетрадки Jupyter в GitHub-репозитории курса.
Как хорошо вы разбираетесь в Data Science? Тест на реальные знания от Tproger и «МегаФон»
8 вопросов-задачек по Data Science. Если ответите правильно больше чем на половину, сможете получить предложение от «МегаФона».
Увлекаетесь Data Science и хотите проверить свои знания? Попробуйте пройти наш тест — 8 вопросов-задачек, которые покажут, разбираетесь ли вы в теме. Это не только тест, но и своеобразный этап отбора: если ответите правильно больше чем на половину вопросов, сможете получить предложение от «МегаФона».

Следите за новыми постами по любимым темам
Подпишитесь на интересующие вас теги, чтобы следить за новыми постами и быть в курсе событий.
Алгоритмы сэмплирования
Сэмплирование (англ. data sampling) — метод корректировки обучающей выборки с целью балансировки распределения классов в исходном наборе данных. Нужно отличать этот метод от сэмплирования в активном обучении для отбора кандидатов и от сэмплирования в статистике [1] для создания подвыборки с сохранением распределения классов.
Неравномерное распределение может быть следующих типов:
- Недостаточное представление класса в независимой переменной;
- Недостаточное представление класса в зависимой переменной.
Многие модели машинного обучения, например, нейронные сети, дают более надежные прогнозы на основе обучения со сбалансированными данными. Однако некоторые аналитические методы, в частности линейная регрессия и логистическая регрессия, не получают дополнительного преимущества.
Когда в обучающем наборе данных доля примеров некоторого класса слишком мала, такие классы называются миноритарными (англ. minority), другие, со слишком большим количеством представителей, — мажоритарными (англ. majority). Подобные тенденции хорошо заметны в кредитном скоринге, в медицине, в директ-маркетинге.
Следует отметить то, что значимость ошибочной классификации может быть разной. Неверная классификация примеров миноритарного класса, как правило, обходится в разы дороже, чем ошибочная классификация примеров мажоритарного класса. Например, при классификации людей, обследованных в больнице, на больных раком (миноритарный класс) и здоровых (мажоритарный класс) лучше будет отправить на дополнительное обследование здоровых пациентов, чем пропустить людей с раком.
Стратегии сэмплирования
- Cубдискретизация (англ. under-sampling) — удаление некоторого количества примеров мажоритарного класса.
- Передискретизации (англ. over-sampling) — увеличение количества примеров миноритарного класса.
- Комбинирование (англ. сombining over- and under-sampling) — последовательное применение субдискретизации и передискретизации.
- Ансамбль сбалансированных наборов (англ. ensemble balanced sets) — использование встроенных методов сэмплирования в процессе построения ансамблей классификаторов.
Также все методы можно разделить на две группы: случайные (недетерминированные) и специальные (детерминированные).
- Случайное сэмплирование (англ. random sampling) — для этого типа сэмплирования существует равная вероятность выбора любого конкретного элемента. Например, выбор 10 чисел в промежутке от 1 до 100. Здесь каждое число имеет равную вероятность быть выбранным.
- Сэплирование с заменой (англ. sampling with replacement) — здесь элемент, который выбирается первым, не должен влиять на вторую или любую другую выборку. Математически, ковариация равна нулю между двумя выборками. Мы должны использовать выборку с заменой, когда у нас большой набор данных. Потому что, если мы используем выборку без замены, то вероятность для каждого предмета, который будет выбран, будет изменяться, и она будет слишком сложной после определенного момента. Выборка с заменой может сказать нам, что чаще встречается в наших данных.
- Сэмплирование без замены (англ. sampling without replacement) — здесь то, что мы выбираем первым, повлияет на второе. Выборка без замены полезна, если набор данных мал. Математически, ковариация между двумя выборками не равна нулю.
Метод Uncertainty Sampling
Идея: выбирать [math]x_i[/math] с наибольшей неопределенностью [math]a(x_i)[/math] .
Задача многоклассовой классификации:
[math]a(x)=\arg\max\limits_
[math]p_k(x), k=1\ldots\left | Y \right |[/math] — ранжированные по убыванию [math]P(y \mid x), y\in Y[/math] .
- Принцип наименьшей достоверности (англ. least confidence):
- Принцип наименьшей разности отступов (англ. margin sampling):
- Принцип максимума энтропии (англ. maximum entropy):
В случае двух классов эти три принципа эквивалентны. В случае многих классов появляются различия.
Примеры алгоритмов
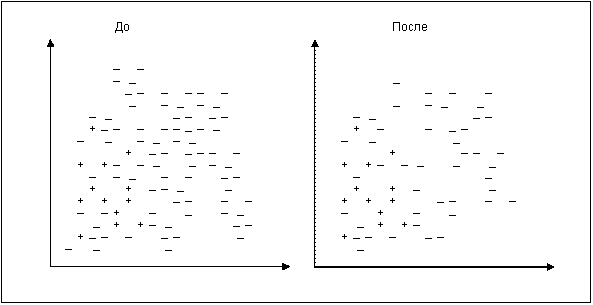
Рис. [math]1[/math] . Случайное удаление примеров мажоритарного класса
Cубдискретизация (удаление примеров мажоритарного класса)
Случайное удаление примеров мажоритарного класса (англ. Random Undersampling)
Это самый простой алгоритм. Рассчитывается число [math]K[/math] – количество мажоритарных примеров, которое необходимо удалить для достижения требуемого уровня соотношения различных классов. Затем случайным образом выбираются K мажоритарных примеров и удаляются. На рис. [math]1[/math] изображены примеры некоторого набора данных в двумерном пространстве признаков до и после использования алгоритма.
Поиск связей Томека (англ. Tomek Links)

Рис. [math]2[/math] . Удаление мажоритарных примеров, участвующих в связях Томека
Пусть примеры [math]E_i[/math] и [math]E_j[/math] принадлежат к различным классам, [math]d(E_i,E_j)[/math] – расстояние между указанными примерами. Пара [math](E_i,E_j)[/math] называется связью Томека, если не найдется ни одного примера [math]E_l[/math] такого, что будет справедлива совокупность неравенств:
[math] \begin d(E_i,E_l)\lt d(E_i,E_j),\\ d(E_j,E_l)\lt d(E_i,E_j) \end [/math]
Согласно данному подходу, все мажоритарные записи, входящие в связи Томека, должны быть удалены из набора данных. Этот способ хорошо удаляет записи, которые можно рассматривать в качестве «зашумляющих». На рис. [math]2[/math] визуально показан набор данных в двумерном пространстве признаков до и после применения стратегии поиска связей Томека.
Правило сосредоточенного ближайшего соседа (англ. Condensed Nearest Neighbor Rule)
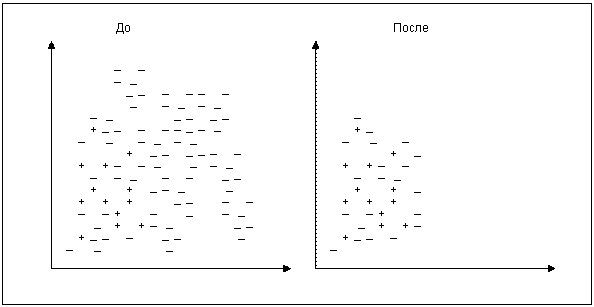
Рис. [math]3[/math] . Удаление примеров мажоритарного класса правилом сосредоточенного ближайшего соседа
Пусть [math]L[/math] – исходный набор данных. Из него выбираются все миноритарные примеры и (случайным образом) один мажоритарный. Обозначим это множество как [math]S[/math] . Все примеры из [math]L[/math] классифицируются по правилу одного ближайшего соседа. Записи, получившие ошибочную метку, добавляются во множество [math]S[/math] (рис. [math]3[/math] ). Таким образом, мы будем учить классификатор находить отличие между похожими примерами, но принадлежащими к разным классам.
Односторонний сэмплинг (англ. One-side sampling, one-sided selection)
Главная идея этой стратегии – это последовательное сочетание предыдущих двух, рассмотренных выше. Для этого на первом шаге применяется правило сосредоточенного ближайшего соседа, а на втором – удаляются все мажоритарные примеры, участвующие в связях Томека. Таким образом, удаляются большие «сгустки» мажоритарных примеров, а затем область пространства со скоплением миноритарных очищается от потенциального шума.
Правило «очищающего» соседа (англ. Neighborhood cleaning rule)
Эта стратегия также направлена на то, чтобы удалить те примеры, которые негативно влияют на исход классификации миноритарных классов. Для этого все примеры классифицируются по правилу трех ближайших соседей. Удаляются следующие мажоритарные примеры:
- получившие верную метку класса;
- являющиеся соседями миноритарных примеров, которые были неверно классифицированы.
Дополнительные
- Under-sampling with Cluster Centroids [2] — уменьшает количество примеров мажоритарного класса, заменяя некоторые кластеры примеров мажоритарного класса их представителем (центроидом кластера).
- NearMiss [math](1 \And 2 \And 3)[/math] [3] — удаляет примеры мажоритарного класса, для которых среднее расстояние до ближайших соседей (KNN) миноритарного класса является наименьшим. Также может использоваться расстояние до самых дальних соседей, либо среднее расстояние до всех соседей.
- Edited Nearest Neighbours [4] — удаляет примеры мажоритарного класса, если при классификации методом KNN они определяются как примеры миноритарного класса.
Передискретизации (увеличение числа примеров миноритарного класса)
Дублирование примеров миноритарного класса (англ. Oversampling)
Самый простой метод – это дублирование примеров миноритарного класса. В зависимости от того, какое соотношение классов необходимо, выбирается количество случайных записей для дублирования.
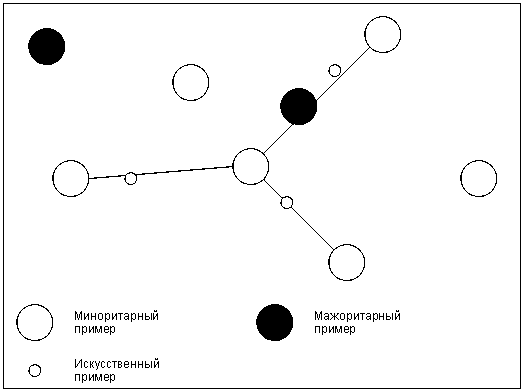
Рис. [math]4[/math] . Искусственно созданные новые примеры миноритарного класса
SMOTE (англ. Synthetic Minority Oversampling Technique)
Этот алгоритм основан на идее генерации некоторого количества искусственных примеров, которые были бы похожи на имеющиеся в миноритарном классе, но при этом не дублировали их. Для создания новой записи находят разность [math]d=X_b–X_a[/math] , где [math]X_a[/math] , [math]X_b[/math] – векторы признаков «соседних» примеров [math]a[/math] и [math]b[/math] из миноритарного класса. Их находят, используя алгоритм ближайшего соседа KNN. В данном случае необходимо и достаточно для примера [math]b[/math] получить набор из [math]k[/math] соседей, из которого в дальнейшем будет выбрана запись [math]b[/math] . Остальные шаги алгоритма KNN не требуются. Далее из [math]d[/math] путем умножения каждого его элемента на случайное число в интервале [math](0, 1)[/math] получают [math]\hat[/math] . Вектор признаков нового примера вычисляется путем сложения [math]X_a[/math] и [math]\hat[/math] . Алгоритм SMOTE позволяет задавать количество записей, которое необходимо искусственно сгенерировать. Степень сходства примеров [math]a[/math] и [math]b[/math] можно регулировать путем изменения числа ближайших соседей [math]k[/math] . На рис. [math]4[/math] схематично изображено то, как в двумерном пространстве признаков могут располагаться искусственно сгенерированные примеры.
В SMOTE (техника избыточной выборки синтетического меньшинства) мы синтезируем элементы для класса меньшинства в непосредственной близости от уже существующих элементов.
from imblearn.over_sampling import SMOTE
smote = SMOTE(ratio=’minority’)
X_sm, y_sm = smote.fit_sample(X, y)
В библиотеке imblearn есть множество других методов как для недостаточной выборки (Cluster Centroids, NearMiss и т.д.), так и для избыточной выборки (ADASYN и bSMOTE).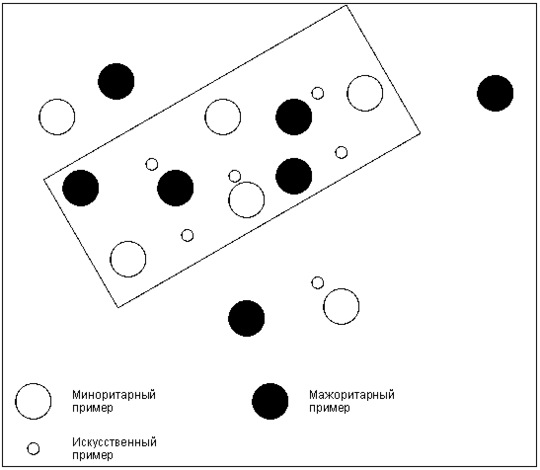
Рис. [math]5[/math] . Негативное влияние алгоритма SMOTE
ASMO (англ. Adaptive Synthetic Minority Oversampling)
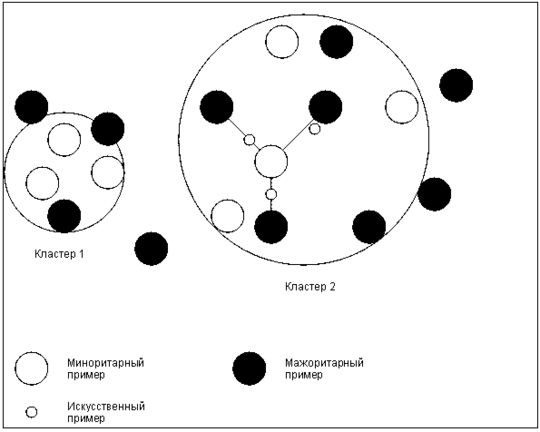
Рис. [math]6[/math] . Основная идея алгоритма ASMO
Алгоритм SMOTE имеет недостаток в том, что «вслепую» увеличивает плотность примерами в области слабо представленного класса (рис. [math]5[/math] ). В случае, если миноритарные примеры равномерно распределены среди мажоритарных и имеют низкую плотность, алгоритм SMOTE только сильнее перемешает классы. В качестве решения данной проблемы был предложен алгоритм адаптивного искусственного увеличения числа примеров миноритарного класса ASMO:
- Если для каждого [math]i[/math] -ого примера миноритарного класса из [math]k[/math] ближайших соседей [math]g, (g≤k)[/math] принадлежит к мажоритарному, то набор данных считается «рассеянным». В этом случае используют алгоритм ASMO, иначе применяют SMOTE (как правило, [math]g[/math] задают равным [math]20[/math] ).
- Используя только примеры миноритарного класса, выделить несколько кластеров (например, алгоритмом [math]\mathrm[/math] -средних).
- Сгенерировать искусственные записи в пределах отдельных кластеров на основе всех классов. Для каждого примера миноритарного класса находят [math]m[/math] ближайших соседей, и на основе них (также как в SMOTE) создаются новые записи.
Такая модификация алгоритма SMOTE делает его более адаптивным к различным наборам данных с несбалансированными классами. Общее представление идеи алгоритма показано на рис. [math]6[/math] .
Дополнительные
- SMOTENC [5] — в отличие от SMOTE, работает с непрерывными признаками у примеров обучающей выборки.
- Borderline-SMOTE [math](1 \And 2)[/math] [6] — в отличие от SMOTE, для создания новых синтетических примеров используются только примеры на границе классов.
- SVM SMOTE — Support Vectors SMOTE [7] — вариант алгоритма SMOTE, который использует алгоритм SVM для обнаружения примеров, рядом с которыми будут создаваться новые синтетические примеры.
Алгоритм Метрополиса — Гастингса
Алгоритм позволяет семплировать любую функцию распределения. Он основан на создании цепи Маркова, то есть на каждом шаге алгоритма новое выбранное значение зависит только от предыдущего.
- Очередная итерация начинается с состояния [math]x^[/math]
- Выбираем [math]x^<\prime>[/math] по распределению [math]q(x^\prime; x^)[/math]
- Вычисляем:
- С вероятностью [math]a(x^<\prime>, x)[/math] ( [math]1[/math] , если [math]a\geq 1[/math] ) [math]x^:=x^<\prime>[/math] , иначе [math]x^:=x^[/math]
Сэмплирование по Гиббсу
Этот алгоритм является частным случаем алгоритма Метрополиса — Гастингса и назван в честь физика Джозайи Гиббса. Он замечателен тем, что для него не требуется явно выраженное совместное распределение, а нужны лишь условные вероятности для каждой переменной, входящей в распределение. Алгоритм на каждом шаге берет одну случайную величину и выбирает её значение при условии фиксированных остальных.
[math]x_^[/math] выбираем по распределению [math]p(x_\mid x_^,\ldots,x_^,x_^,\ldots,x_^)[/math] и повторяем.
Это частный случай алгоритма Метрополиса для распределений [math]q(x^<\prime>; x)=p(x_^<\prime>\mid x_)[/math] , и вероятность принятия каждого сэмпла полается равна [math]1[/math] . Поэтому сэмплирование по Гиббсу сходится, и, так как это такое же случайное блуждание по сути, верна та же квадратичная оценка. В больших размерностях может оказаться эффективнее сэмплить по несколько переменных сразу, а не по одной — например, часто бывает, что у нас двудольный граф из переменных, в которых все переменные из одной доли связаны со всеми переменными из другой доли (ну или со многими), а между собой не связаны. В такой ситуации следует зафиксировать все переменные одной доли и просэмплировать все переменные в другой доле одновременно (это можно понимать буквально — поскольку при такой структуре все переменные одной доли условно независимы при условии другой, их можно сэмплировать независимо и параллельно), потом зафиксировать все переменные второй доли и так далее.Slice sampling
Выборка среза представляет собой тип алгоритма Монте Карло по схеме марковских цепей для выборки псевдослучайных чисел, т.е. для отбора случайных выборок из статистического распределения. Метод основан на наблюдении, что для выборки случайной величины можно равномерно выбирать из области под графиком ее функции плотности.
Slice sampling, в его самой простой форме, равномерно выбирается из-под кривой [math]f(x)[/math] без необходимости отбрасывать какие-либо точки следующими действиями:
- Выберите начальное значение [math]x_[/math] , для которого [math]f(x_)\gt 0[/math]
- Выберите значение [math]y[/math] равномерно между [math]0[/math] и [math]f(x_)[/math]
- Проведите горизонтальную линию через кривую в этой координате [math]y[/math]
- Выберите точку [math](x, y)[/math] на отрезке в пределах кривой
- Повторите с шага [math]2[/math] , используя новое значение [math]x[/math]
Суть здесь заключается в том, что один из способов равномерной выборки точки из произвольной кривой — это сначала нарисовать тонкие горизонтальные срезы одинаковой высоты по всей кривой. Затем мы можем сэмплировать точку внутри кривой путем случайного выбора среза, который находится в точке или ниже кривой в позиции [math]x[/math] на предыдущей итерации, а затем случайным образом выбрать позицию [math]x[/math] где-нибудь вдоль среза. Используя позицию [math]x[/math] из предыдущей итерации алгоритма, в долгосрочной перспективе мы выбираем срезы с вероятностями, пропорциональными длине их сегментов в пределах кривой. Самая сложная часть этого алгоритма — это поиск границ горизонтального среза, который включает в себя инвертирование функции, описывающей распределение, из которого производится выборка. Это особенно проблематично для мультимодальных распределений, где срез может состоять из нескольких прерывистых частей. Часто можно использовать форму выборки отклонения, чтобы преодолеть это, когда мы производим выборку из более крупного среза, который, как известно, включает в себя требуемый рассматриваемый срез, а затем отбрасываем точки за пределами желаемого среза. Этот алгоритм можно использовать для выборки из области под любой кривой, независимо от того, интегрируется ли функция в [math]1[/math] . Фактически, масштабирование функции по константе не влияет на выборочные [math]x[/math] —позиции. Это означает, что алгоритм может использоваться для выборки из распределения, функция плотности вероятности которого известна только с точностью до константы.
Комбинирование
- SMOTE [math]+[/math] Tomek links [8] — сначала выполняет передискретизацию с использованием SMOTE, а потом субдискретизацию используя Tomek Links.
- SMOTE [math]+[/math] ENN [9] — последовательно использует SMOTE и Edited Nearest Neighbours.
Ансамбль сбалансированных наборов
- Easy Ensemble classifier [10] — независимые классификаторы обучаются на случайных подвыборках, из которых постепенно удаляются правильно классифицирующиеся примеры мажоритарных классов.
- Balanced Random Forest [11] — в отличие от классического случайного леса, может работать на несбалансированных данных.
- Balanced Bagging [12] — в отличие от классического бэггинга, имеет дополнительный шаг субдискретизации обучающей подвыборки.
Реализации
Imbalanced-learn — набор инструментов с открытым исходным кодом на Python, целью которого является предоставление широкого спектра методов для решения проблемы несбалансированного набора данных. На рис. [math]7[/math] представлена таблица реализованных в библиотеке методов.
Пример кода для передискретизации набора данных с использованием SMOTE:
from sklearn.datasets import make_classification from sklearn.decomposition import PCA from imblearn.oversampling import SMOTE # Создание датасета X, y = makeclassification (n_classes=2, weights =[0.1, 0.9], n_features=20, n_samples=5000) Применение SMOTE over-sampling sm = SMOTE(ratio=’auto’, kind=’regular’) X_resampled , y_resampled=sm.fit_sample(X, y)

Рис. [math]7[/math] . Методы imbalanced-learn
См. также
- Метрический классификатор и метод ближайших соседей
- Байесовская классификация
- Активное обучение
- Виды ансамблей
Примечания
- ↑Sampling (statistics)
- ↑Show-Jane Yen, Yue-Shi Lee,Cluster-based under-sampling approaches for imbalanced data distributions, Expert Systems with Applications, Volume 36, Issue 3, Part 1, 2009, Pages 5718-5727, ISSN 0957-4174
- ↑ I. Mani, J. Zhang. “kNN approach to unbalanced data distributions: A case study involving information extraction,” In Proceedings of the Workshop on Learning from Imbalanced Data Sets, pp. 1-7, 2003.
- ↑ D. Wilson, “Asymptotic Properties of Nearest Neighbor Rules Using Edited Data,” IEEE Transactions on Systems, Man, and Cybernetrics, vol. 2(3), pp. 408-421, 1972.
- ↑ N. V. Chawla, K. W. Bowyer, L. O. Hall, W. P. Kegelmeyer, “SMOTE: Synthetic minority over-sampling technique,” Journal of Artificial Intelligence Research, vol. 16, pp. 321-357, 2002.
- ↑ H. Han, W.-Y. Wang, B.-H. Mao, “Borderline-SMOTE: A new over-sampling method in imbalanced data sets learning,” In Proceedings of the 1st International Conference on Intelligent Computing, pp. 878-887, 2005.
- ↑ H. M. Nguyen, E. W. Cooper, K. Kamei, “Borderline over-sampling for imbalanced data classification,” In Proceedings of the 5th International Workshop on computational Intelligence and Applications, pp. 24-29, 2009.
- ↑ G. E. A. P. A. Batista, A. L. C. Bazzan, M. C. Monard, “Balancing training data for automated annotation of keywords: A case study,” In Proceedings of the 2nd Brazilian Workshop on Bioinformatics, pp. 10-18, 2003.
- ↑ G. E. A. P. A. Batista, R. C. Prati, M. C. Monard, “A study of the behavior of several methods for balancing machine learning training data,” ACM Sigkdd Explorations Newsletter, vol. 6(1), pp. 20-29, 2004.
- ↑ X.-Y. Liu, J. Wu and Z.-H. Zhou, “Exploratory undersampling for class-imbalance learning,” IEEE Transactions on Systems, Man, and Cybernetics, vol. 39(2), pp. 539-550, 2009.
- ↑ C. Chao, A. Liaw, and L. Breiman. «Using random forest to learn imbalanced data.» University of California, Berkeley 110 (2004): 1-12.
- ↑ Hido, Shohei & Kashima, Hisashi. (2008). Roughly Balanced Bagging for Imbalanced Data. 143-152. 10.1137/1.9781611972788.13.
Источники информации
- Oversampling and undersampling in data analysis
- Различные стратегии сэмплинга в условиях несбалансированности классов
- Lemaître, G. Nogueira, F. Aridas, Ch.K. (2017) Imbalanced-learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning, Journal of Machine Learning Research, vol. 18, no. 17, 2017, pp. 1-5.
- Машинное обучение
- Классификация и регрессия